Las superficies metálicas ayudan a la conmutación cuántica molecular
Los interruptores moleculares cuánticos funcionan por medio de túneles de hidrógeno con algo de ayuda
La dinámica cuántica del hidrógeno es fundamental para muchos problemas de la naturaleza, ya que está fuertemente influenciada por el entorno en el que se produce. En su contribución a la PRL, los miembros del Grupo de Lise Meitner en la MPSD abordan la transferencia de hidrógeno dentro de un interruptor molecular apoyado, demostrando que el apoyo de la superficie puede desempeñar un papel decisivo en la reacción de tunelización.
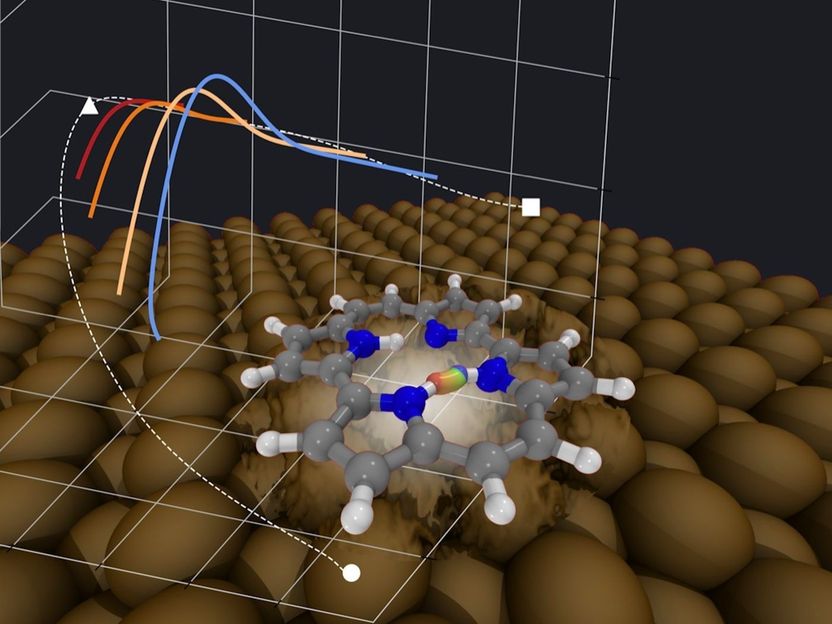
Dentro de la molécula de porfirio adsorbida en las superficies metálicas fcc[110], las reacciones de transferencia de hidrógeno se producen a través de túneles nucleares, incluso justo por debajo de la temperatura ambiente. La figura muestra esquemáticamente una ruta de tunelización instantánea, obtenida en una superficie de energía potencial de principios de primera dimensión completa, en el espacio real y proyectada en coordenadas moleculares seleccionadas. La fluctuación de los átomos de la superficie puede aumentar la tasa de tunelización en unos pocos órdenes de magnitud.
© Mariana Rossi
En la constante búsqueda de miniaturizar los componentes tecnológicos, la nanotecnología basada en las moléculas se convierte en un campo de interés cada vez mayor. En este campo, las moléculas individuales se convierten en los componentes fundamentales de los dispositivos electrónicos. La gran variedad de arquitecturas moleculares posibles y la posibilidad de manipular con precisión la síntesis molecular abre la puerta a un sinfín de componentes funcionales. Sin embargo, el desafío clave es conseguir el control de estas funciones en la nanoescala, donde los efectos de la mecánica cuántica se hacen prominentes.
La molécula de porfirio es un ejemplo de un prototipo de interruptor molecular. El porficeno es un isómero estructural de la porfirina con fuertes enlaces H en su cavidad interna. Su capacidad de conmutación se basa en una reacción muy fundamental en la física química: una doble transferencia de hidrógeno que puede intercambiar las posiciones de los hidrógenos en la cavidad interna y así definir diferentes estados (on/off) de la molécula - un proceso llamado tautomerización. Para controlar y medir la estructura atómica y la velocidad de conmutación de estas unidades moleculares, normalmente se inmovilizan poniéndolas en contacto con superficies metálicas. Esta situación exige que los investigadores comprendan la dinámica del hidrógeno dentro de un entorno que incluye tipos de interacción cualitativamente diferentes entre los átomos dentro de la molécula y entre la molécula y la superficie.
En este contexto, el porfíceno ha sido ampliamente estudiado mediante técnicas experimentales de una sola molécula. Los investigadores han observado varios aspectos desconcertantes de la tasa de tautomerización en diferentes rangos de temperatura, incluidas las temperaturas en las que los átomos ya no se comportan como las partículas clásicas, sino que pueden hacer túneles a través de las barreras. Usando una montaña como analogía, los átomos viajarían instantáneamente entre dos valles en línea recta por debajo de la montaña, en lugar de tomarse el tiempo de subir y bajar por encima de ella.
En su nuevo trabajo recién publicado en PRL, Yair Litman y Mariana Rossi abordan este cambio molecular apoyado con la metodología más avanzada y nuevos algoritmos de computación: una combinación de la teoría funcional de la densidad con instantáneos de polímeros de anillo. Estos métodos permitieron finalmente estudiar tales sistemas con simulaciones atomísticas a escala completa que tratan tanto a los electrones como a los núcleos como partículas de mecánica cuántica. Los autores muestran que, en el caso del porficeno adsorbido en superficies de Cu(110) y Ag(110), la reacción de transferencia de hidrógeno presenta, en efecto, una gran contribución de los túneles nucleares incluso a temperaturas no muy inferiores a la temperatura ambiente.
De manera bastante sorprendente, los autores descubrieron que con la disminución de la temperatura, los átomos pesados de la superficie, como el cobre, participan en la reacción de tunelización intramolecular de hidrógeno y pueden causar un aumento de la tasa de tunelización de hasta dos órdenes de magnitud a una temperatura de alrededor de 80 K. Cuanto más fuerte sea la interacción de la molécula con la superficie (hibridación de orbitales electrónicos), más pronunciada será la participación de los átomos de la superficie en el evento de tunelización.
En particular, los autores también explicaron una dependencia no convencional de la temperatura de la tasa de tunelización, que se había observado anteriormente en los experimentos. Se debe a la existencia de una estructura metaestable intermedia en la reacción, que existe durante un período de tiempo tan corto (~100 picosegundos, siendo un picosegundo una trillonésima de segundo) que no pudo ser detectada por las técnicas experimentales empleadas anteriormente en este sistema. Comprendiendo este mecanismo, los autores también podrían explicar los diferentes regímenes de dependencia de la temperatura de la tasa en el régimen de túneles y proponer un modelo simple para predecir esta dependencia de la temperatura para este interruptor adsorbido en otras superficies metálicas.
Se trata de nuevos e importantes conocimientos sobre el hecho de que ciertas características del soporte de la superficie pueden influir en las propiedades mecánicas cuánticas nucleares de la reacción de conmutación en estas, y probablemente en otras, moléculas. También demuestran que los sustratos monocristalinos son una plataforma ideal en la que la teoría de vanguardia y el experimento pueden unirse para proporcionar una comprensión más profunda de la dinámica cuántica nuclear en entornos complejos. Tales hallazgos tienen una importancia fundamental considerable y pueden también guiar el diseño e interpretación de las arquitecturas experimentales en el desarrollo de la nanotecnología molecular.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Más noticias del departamento ciencias



















































