Investigación de catalizadores: Las sondas moleculares requieren cálculos muy precisos
Los científicos utilizan métodos avanzados con funciones híbridas para el análisis de sitios activos
Los catalizadores son indispensables para muchas tecnologías. Para mejorar aún más los catalizadores heterogéneos, es necesario analizar los complejos procesos en sus superficies, donde se encuentran los sitios activos. Los científicos del Instituto Tecnológico de Karlsruhe (KIT), junto con sus colegas de España y Argentina, han alcanzado un progreso decisivo: Como se informa en las Physical Review Letters, utilizan métodos de cálculo con las llamadas funciones híbridas para la interpretación fiable de los datos experimentales.
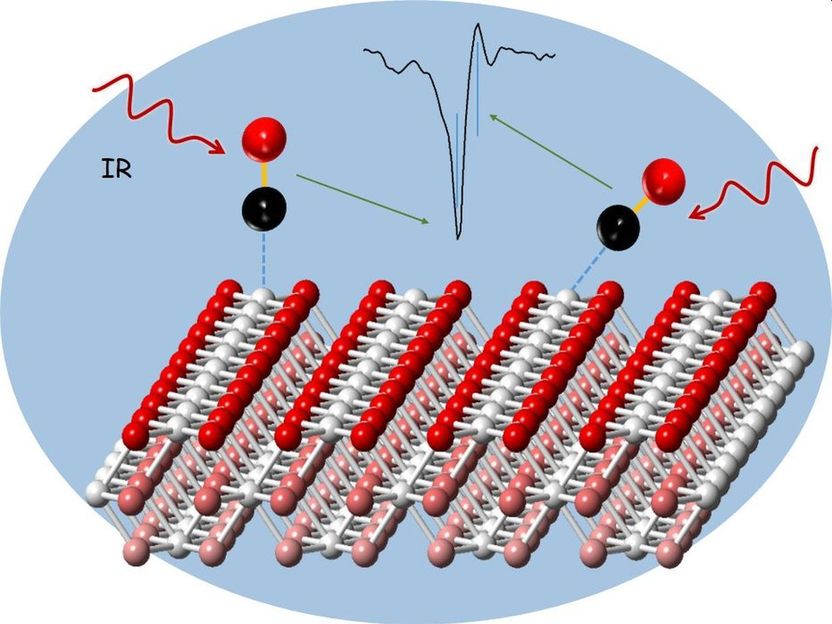
Análisis de un catalizador de óxido de cerio utilizando moléculas de sonda de monóxido de carbono y espectroscopia de absorción por reflexión infrarroja.
IFG/KIT
Muchas tecnologías importantes, como los procesos de conversión de energía, la reducción de emisiones o la producción de productos químicos, funcionan únicamente con catalizadores adecuados. Por esta razón, los materiales de alta eficiencia para la catálisis heterogénea están ganando importancia. En la catálisis heterogénea, el material que actúa como catalizador y las sustancias que reaccionan existen en diferentes fases como, por ejemplo, un sólido o un gas. La composición de los materiales puede determinarse de manera fiable mediante diversos métodos. Sin embargo, los procesos que tienen lugar en la superficie del catalizador no pueden detectarse con casi ningún método de análisis. "Pero son estos procesos químicos altamente complejos en la superficie más exterior del catalizador los que tienen una importancia decisiva", dice el Profesor Christof Wöll, Jefe del Instituto de Interfaces Funcionales (IFG) del KIT. "Allí se encuentran los sitios activos, donde tiene lugar la reacción catalizada".
Examen preciso de la superficie de los catalizadores en polvo
Entre los catalizadores heterogéneos más importantes se encuentran los óxidos de cerio, es decir, los compuestos del cerio metálico de tierras raras con oxígeno. Existen en forma de polvo y consisten en nanopartículas de estructura controlada. La forma de las nanopartículas influye considerablemente en la reactividad del catalizador. Para estudiar los procesos en la superficie de esos catalizadores en polvo, los investigadores han empezado recientemente a utilizar moléculas de sonda, como las de monóxido de carbono, que se unen a las nanopartículas. Estas sondas se miden entonces por espectroscopia de absorción de reflexión infrarroja (IRRAS). La radiación infrarroja hace que las moléculas vibren. A partir de las frecuencias de vibración de las moléculas de las sondas, se puede obtener información detallada sobre el tipo y la composición de los sitios catalíticos. Hasta ahora, sin embargo, la interpretación de los datos experimentales de la IRRAS ha sido muy difícil, porque los catalizadores en polvo tecnológicamente relevantes tienen muchas bandas de vibración, cuya asignación exacta es un desafío. Los cálculos teóricos no han sido de ninguna ayuda, porque la desviación del experimento, también en el caso de los sistemas modelo, era tan grande que las bandas de vibración observadas experimentalmente no podían asignarse con precisión.
Largo tiempo de cálculo - Alta precisión
Los investigadores del Instituto de Interfaces Funcionales (IFG) y del Instituto de Investigación y Tecnología de Catálisis (IKFT) del KIT, en cooperación con colegas de España y Argentina coordinados por la Dra. M. Verónica Ganduglia-Pirovano del Consejo Superior de Investigaciones Científicas (CSIC) de Madrid, han identificado y resuelto un importante problema de análisis teórico. Como se informó en las cartas de revisión física, los estudios teóricos sistemáticos y la validación de los resultados mediante sistemas de modelos revelaron que los métodos teóricos utilizados hasta ahora tienen algunas debilidades fundamentales. En general, esas debilidades pueden observarse en los cálculos que utilizan la teoría funcional de la densidad (DFT), un método con el que se puede determinar el estado básico de la mecánica cuántica de un sistema multielectrónico en función de la densidad de los electrones. Los investigadores descubrieron que las debilidades pueden superarse con las llamadas funciones híbridas que combinan la DFT con el método Hartree-Fock, un método de aproximación en la química cuántica. Esto hace que los cálculos sean muy complejos, pero también muy precisos. "Los tiempos de cálculo requeridos por estos nuevos métodos son más largos por un factor de 100 que para los métodos convencionales", dice Christof Wöll. "Pero este inconveniente está más que compensado por el excelente acuerdo con los sistemas experimentales". Utilizando catalizadores de óxido de cerio a nanoescala, los investigadores demostraron este progreso que puede contribuir a que los catalizadores heterogéneos sean más eficaces y duraderos.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias





















Noticias más leídas











Más noticias de nuestros otros portales




















Contenido visto recientemente
New-York Hamburger Gummi-Waaren Compagnie AG - Lüneburg, Alemania
