El superordenador y las simulaciones cuánticas resuelven un difícil problema de la ciencia de los materiales
Investigadores japoneses calculan con éxito la energía de flexión de la molécula de disiloxano con simulaciones moleculares ultrafinas realizadas en un superordenador
Comprender las propiedades estructurales de las moléculas que se encuentran en la naturaleza o se sintetizan en el laboratorio siempre ha sido el pan de cada día de los científicos de materiales. Pero, con los avances de la ciencia y la tecnología, la tarea se ha vuelto aún más ambiciosa: descubrir nuevos materiales con propiedades muy deseables. Para llevar a cabo esta hazaña de forma sistemática, los científicos de materiales recurren a sofisticadas técnicas de simulación que incorporan las reglas de la mecánica cuántica, las mismas que rigen las propias moléculas.
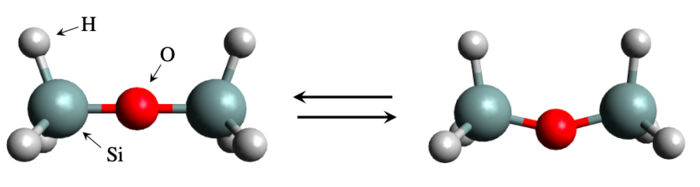
Estructura de la molécula de disiloxano en forma lineal (izquierda) y en forma doblada (derecha). Un equipo mundial de científicos estudió la energía de flexión de una molécula de silicato, el disiloxano. Aunque el sistema parece sencillo y manejable, el cálculo de la energía de flexión es en realidad un problema difícil de resolver con los métodos de simulación convencionales.
Dr. Kenta Hongo from JAIST
El enfoque basado en la simulación ha tenido un éxito notable, hasta el punto de que se le ha dedicado todo un campo de estudio llamado informática de materiales. Pero también ha habido casos de fracaso. Un ejemplo notable es el del disiloxano, un compuesto de silicio (Si) formado por un puente Si-O-Si con tres átomos de hidrógeno en cada extremo. La estructura es bastante sencilla y, sin embargo, ha sido notoriamente difícil estimar cuánta energía se necesita para doblar el puente Si-O-Si. Los resultados experimentales han sido inconsistentes y los cálculos teóricos han arrojado valores muy diferentes debido a la sensibilidad de las propiedades calculadas a la elección de los parámetros y al nivel de la teoría.
Afortunadamente, un equipo de investigación internacional dirigido por el Dr. Kenta Hongo, profesor asociado del Instituto Avanzado de Ciencia y Tecnología de Japón, ha conseguido resolver este problema. En su estudio, publicado en Physical Chemistry Chemical Physics, el equipo logró esta hazaña utilizando una técnica de simulación de última generación denominada "método de Monte Carlo cuántico de primeros principios", que finalmente superó las dificultades que otras técnicas estándar no pudieron superar.
¿Pero todo se reduce a mejores simulaciones? No del todo. "Obtener una respuesta que no coincide con el valor conocido experimentalmente no es, en sí mismo, sorprendente. La concordancia puede mejorar con simulaciones más cuidadosas y costosas. Pero en el caso del disiloxano, la concordancia empeora con simulaciones más cuidadosas", explica el Dr. Hongo. "Lo que nuestro método ha conseguido, más bien, son buenos resultados sin mucha dependencia de los parámetros de ajuste, de modo que no tenemos que preocuparnos de si los valores ajustados son suficientes".
El equipo comparó el enfoque de Monte Carlo cuántico de primeros principios con otras técnicas estándar, como los cálculos de la "teoría funcional de la densidad" (DFT) y el "método de clústeres acoplados con sustituciones simples y dobles y triples no iterativos" (CCSD(T)), junto con mediciones empíricas de estudios anteriores. Los tres métodos diferían principalmente en su sensibilidad hacia la "integridad" de los conjuntos de bases (un conjunto de funciones utilizadas para definir las funciones de onda cuánticas).
Resultó que para la DFT y el CCSD(T), la elección del conjunto de bases afectaba a la amplitud y a las posiciones de amplitud cero de las funciones de onda, mientras que para el Monte Carlo cuántico sólo afectaba a las posiciones de amplitud cero. Esto permitía ajustar la amplitud de forma que la forma de la función de onda se aproximara a la de una solución exacta. "Esta propiedad de autocuración de la amplitud funciona bien para reducir la dependencia del conjunto de bases y disminuir el sesgo derivado de un conjunto de bases incompleto al calcular la barrera de energía de flexión", explica el Dr. Hongo.
Aunque se trata de un avance emocionante en sí mismo, el profesor Hongo señala el panorama general. "Las simulaciones moleculares se utilizan ampliamente para diseñar nuevos medicamentos y catalizadores. Deshacerse de las dificultades fundamentales de su uso contribuye en gran medida al diseño de dichos materiales. Con nuestros potentes superordenadores, el método utilizado en nuestro estudio podría ser una estrategia estándar para superar dichas dificultades", afirma.
Esto sí que merece el calificativo de "salto cuántico".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias



















































