Un programa de aprendizaje automático para juegos inspira el desarrollo de una innovadora herramienta científica
Una nueva herramienta de IA modela en tiempo récord el comportamiento de grupos de nanopartículas
Aprendemos nuevas habilidades mediante la repetición y el aprendizaje por refuerzo. Mediante el método de ensayo y error, repetimos acciones que conducen a buenos resultados, tratamos de evitar los malos y buscamos mejorar los intermedios. Los investigadores diseñan ahora algoritmos basados en una forma de inteligencia artificial que utiliza el aprendizaje por refuerzo. Los están aplicando para automatizar la síntesis química, el descubrimiento de fármacos e incluso para jugar a juegos como el ajedrez y el Go.
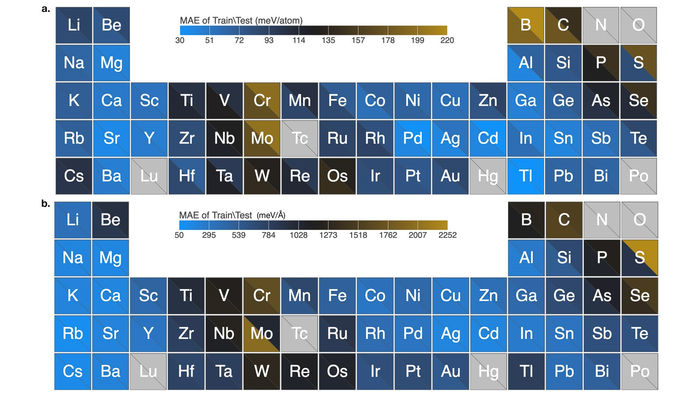
El gráfico muestra el excelente rendimiento del algoritmo para las predicciones del campo de fuerza de los nanoclusters elementales que cubren 54 elementos de la tabla periódica. Los datos demuestran un error absoluto medio muy bajo.
Image by Argonne National Laboratory
Científicos del Laboratorio Nacional de Argonne del Departamento de Energía de EE.UU. (DOE) han desarrollado un algoritmo de aprendizaje por refuerzo para otra aplicación. Se trata de modelar las propiedades de los materiales a escala atómica y molecular y debería acelerar enormemente el descubrimiento de materiales.
Al igual que los humanos, este algoritmo "aprende" a resolver problemas a partir de sus errores y aciertos. Pero lo hace sin intervención humana.
Históricamente, Argonne ha sido un líder mundial en el modelado molecular. Se trata de calcular las fuerzas entre los átomos de un material y utilizar esos datos para simular su comportamiento en diferentes condiciones a lo largo del tiempo.
Sin embargo, los modelos anteriores se basaban en gran medida en la intuición y la experiencia humanas y a menudo requerían años de minuciosos esfuerzos. El algoritmo de aprendizaje por refuerzo del equipo reduce el tiempo a días y horas. Además, produce datos de mayor calidad que los posibles con los métodos convencionales.
"Nuestra inspiración fue AlphaGo", dijo Sukriti Manna, asistente de investigación en el Centro de Materiales a Nanoescala (CNM) de Argonne, una instalación usuaria de la Oficina de Ciencia del DOE. "Es el primer programa informático que derrota a un jugador de Go campeón del mundo".
El tablero estándar de Go tiene 361 casillas posicionales, mucho más grandes que las 64 de un tablero de ajedrez. Eso se traduce en un enorme número de configuraciones posibles del tablero. La clave para que AlphaGo se convirtiera en campeón del mundo fue su capacidad para mejorar sus habilidades mediante el aprendizaje por refuerzo.
La automatización del modelado molecular es, por supuesto, muy diferente a la de un programa de ordenador de Go. "Uno de los retos a los que nos enfrentamos es similar al desarrollo del algoritmo necesario para los coches de autoconducción", afirma Subramanian Sankaranarayanan, jefe de grupo del CNM de Argonne y profesor asociado de la Universidad de Illinois Chicago.
Mientras que el tablero de Go es estático, los entornos de tráfico cambian continuamente. El coche autodirigido tiene que interactuar con otros coches, rutas variadas, señales de tráfico, peatones, intersecciones, etc. Los parámetros relacionados con la toma de decisiones cambian constantemente con el tiempo.
La resolución de problemas difíciles del mundo real en el descubrimiento y diseño de materiales implica también una toma de decisiones continua en la búsqueda de soluciones óptimas. El algoritmo del equipo incluye árboles de decisión que ofrecen un refuerzo positivo en función del grado de éxito en la optimización de los parámetros del modelo. El resultado es un modelo que puede calcular con precisión las propiedades de los materiales y sus cambios a lo largo del tiempo.
El equipo probó con éxito su algoritmo con 54 elementos de la tabla periódica. Su algoritmo aprendió a calcular los campos de fuerza de miles de nanoclusters para cada elemento y realizó los cálculos en un tiempo récord. Estos nanoclusters son conocidos por su compleja química y la dificultad que tienen los métodos tradicionales para modelarlos con precisión.
"Esto es algo parecido a completar los cálculos de varias tesis doctorales en cuestión de días cada una, en lugar de años", dijo Rohit Batra, experto del CNM en herramientas de aprendizaje automático y basado en datos. El equipo realizó estos cálculos no sólo para nanoclusters de un solo elemento, sino también para aleaciones de dos elementos.
"Nuestro trabajo representa un gran paso adelante en este tipo de desarrollo de modelos para la ciencia de los materiales", dijo Sankaranarayanan. "La calidad de nuestros cálculos para los 54 elementos con el algoritmo es muy superior al estado del arte".
La ejecución del algoritmo del equipo requirió cálculos con grandes conjuntos de datos en ordenadores de alto rendimiento. Para ello, el equipo recurrió al clúster de ordenadores de carbono del CNM y al superordenador Theta de la Argonne Leadership Computing Facility, una instalación usuaria de la Oficina de Ciencia del DOE. También recurrieron a los recursos informáticos del National Energy Research Scientific Computing Center, una instalación de la Oficina de Ciencia del DOE en el Lawrence Berkeley National Laboratory.
"El algoritmo debería acelerar en gran medida el tiempo necesario para abordar los grandes retos en muchas áreas de la ciencia de los materiales", dijo Troy Loeffler, químico computacional y teórico del CNM. Los ejemplos incluyen materiales para dispositivos electrónicos, catalizadores para procesos industriales y componentes de baterías.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias




















































