Examen de los desplazamientos atómicos en aleaciones de alta entropía
Las aleaciones de alta entropía están en discusión para aplicaciones muy diferentes
Las aleaciones de alta entropía de metales 3d tienen propiedades intrigantes que resultan interesantes para aplicaciones en el sector energético. Un equipo internacional de BESSY II ha investigado ahora el orden local a escala atómica en una aleación Cantor de alta entropía de cromo, manganeso, hierro, cobalto y níquel. Los resultados de la combinación de estudios espectroscópicos y simulaciones estadísticas amplían la comprensión de este grupo de materiales.
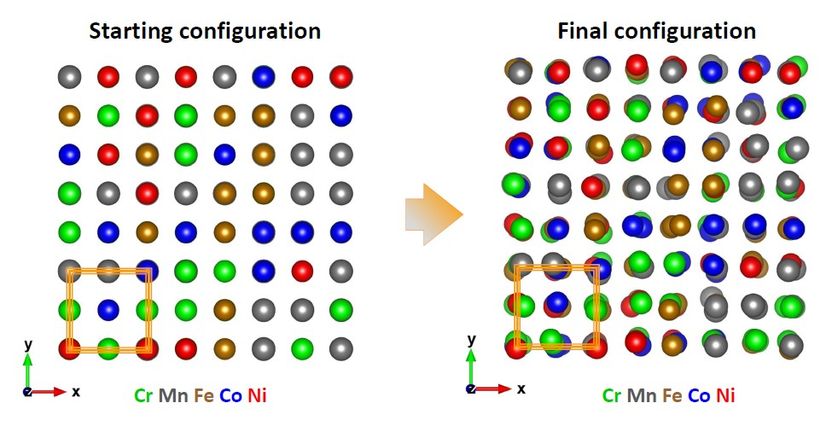
La supercélula se rellena aleatoriamente con los cinco elementos en las posiciones de la red fcc; en la configuración inicial, todas las capas están precisamente superpuestas. Los desplazamientos de todos los elementos en la configuración final se han revelado mediante un ajuste simultáneo de los espectros experimentales independientes con un uso de simulaciones de Monte Carlo inverso.
© A.Kuzmin / University of Latvia and A. Smekhova / HZB
Las aleaciones de alta entropía son objeto de debate para aplicaciones muy diversas: Algunos materiales de este grupo son adecuados para el almacenamiento de hidrógeno, otros para la electrocatálisis sin metales nobles, el blindaje contra la radiación o como supercondensadores.
La estructura microscópica de las aleaciones de alta entropía es muy diversa y cambiante; en particular, el ordenamiento local y la presencia de diferentes fases secundarias afectan significativamente a las propiedades macroscópicas, como la dureza, la resistencia a la corrosión y también el magnetismo. La llamada aleación Cantor, que consta de los elementos cromo, manganeso, hierro, cobalto y níquel mezclados en una proporción equimolar, puede considerarse un sistema modelo adecuado para toda la clase de estos materiales.
Estructura local estudiada en BESSY II
Científicos del Instituto Federal de Investigación de Materiales (BAM, Berlín), la Universidad de Letonia en Riga, Letonia, la Universidad del Ruhr en Bochum y el HZB han estudiado ahora la estructura local de este sistema modelo en detalle. Utilizando la espectroscopia de absorción de rayos X (EXAFS) en BESSY II, han podido rastrear con precisión cada elemento individual y sus desplazamientos desde las posiciones ideales de la red para este sistema de la manera más insesgada con la ayuda de cálculos estadísticos y el método Monte Carlo inverso.
El cromo muestra mayores desplazamientos
De este modo, descubrieron las peculiaridades del entorno local de cada elemento: A pesar de que los cinco elementos de la aleación se distribuyen en los nodos de la red cúbica centrada en la cara y tienen distancias interatómicas promediadas estadísticamente muy cercanas (2,54 - 2,55 Å) con sus vecinos más próximos, se encontraron relajaciones estructurales mayores sólo para los átomos de cromo. Además, no se detectó ninguna evidencia de fases secundarias a escala atómica. Las propiedades magnéticas macroscópicas estudiadas con magnetometría convencional en el HZB CoreLab se correlacionaron con las relajaciones estructurales reveladas del cromo.
"Los resultados describen la disposición de los átomos individuales a escala atómica y cómo puede producirse el complejo orden magnético que revelamos", explica la doctora Alevtina Smekhova, física del HZB, que supervisó los experimentos en el HZB.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias



















































