El aprendizaje automático lleva el modelado de materiales a una nueva era
El aprendizaje profundo permite calcular con precisión la estructura electrónica a gran escala
Anuncios



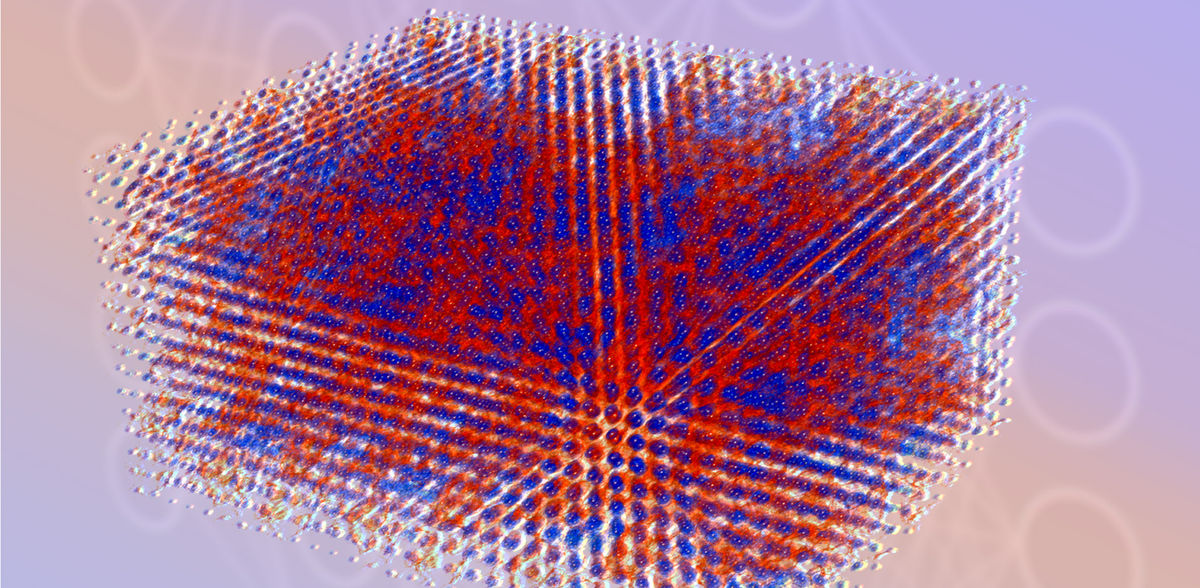
La disposición de los electrones en la materia, conocida como estructura electrónica, desempeña un papel crucial en la investigación fundamental, pero también aplicada, como el diseño de fármacos y el almacenamiento de energía. Sin embargo, la falta de una técnica de simulación que ofrezca tanto alta fidelidad como escalabilidad a través de diferentes escalas de tiempo y longitud ha sido durante mucho tiempo un obstáculo para el progreso de estas tecnologías. Investigadores del Center for Advanced Systems Understanding (CASUS) del Helmholtz-Zentrum Dresden-Rossendorf (HZDR) de Görlitz (Alemania) y del Sandia National Laboratories de Albuquerque (Nuevo México, EE.UU.) son ahora pioneros en un método de simulación basado en el aprendizaje automático(npj Computational Materials) que sustituye a las técnicas tradicionales de simulación de estructuras electrónicas. Su pila de software Materials Learning Algorithms (MALA) permite acceder a escalas de longitud antes inalcanzables.
Los electrones son partículas elementales de importancia fundamental. Sus interacciones mecánicas cuánticas entre sí y con los núcleos atómicos dan lugar a multitud de fenómenos observados en química y ciencia de materiales. Entender y controlar la estructura electrónica de la materia permite comprender mejor la reactividad de las moléculas, la estructura y el transporte de energía dentro de los planetas y los mecanismos de fallo de los materiales.
Los retos científicos se abordan cada vez más mediante el modelado y la simulación computacionales, aprovechando las capacidades de la informática de alto rendimiento. Sin embargo, un obstáculo importante para lograr simulaciones realistas con precisión cuántica es la falta de una técnica de modelización predictiva que combine una gran precisión con la escalabilidad a través de diferentes escalas de longitud y tiempo. Los métodos clásicos de simulación atomística pueden manejar sistemas grandes y complejos, pero su omisión de la estructura electrónica cuántica restringe su aplicabilidad. Por el contrario, los métodos de simulación que no se basan en suposiciones, como el modelado empírico y el ajuste de parámetros (métodos de primeros principios), ofrecen una gran fidelidad, pero son muy exigentes desde el punto de vista computacional. Por ejemplo, la teoría funcional de la densidad (DFT), un método de primeros principios ampliamente utilizado, exhibe un escalado cúbico con el tamaño del sistema, lo que restringe sus capacidades de predicción a escalas pequeñas.

Enfoque híbrido basado en el aprendizaje profundo
El equipo de investigadores ha presentado ahora un novedoso método de simulación denominado pila de software de algoritmos de aprendizaje de materiales (MALA). En informática, una pila de software es una colección de algoritmos y componentes de software que se combinan para crear una aplicación informática para resolver un problema concreto. Lenz Fiedler, estudiante de doctorado y desarrollador clave de MALA en CASUS, explica: "MALA integra el aprendizaje automático con enfoques basados en la física para predecir la estructura electrónica de los materiales. Emplea un enfoque híbrido, utilizando un método de aprendizaje automático establecido llamado aprendizaje profundo para predecir con precisión las cantidades locales, complementado con algoritmos de física para calcular las cantidades globales de interés."
La pila de software de MALA toma como entrada la disposición de los átomos en el espacio y genera huellas digitales conocidas como componentes del bisespectro, que codifican la disposición espacial de los átomos alrededor de un punto de la cuadrícula cartesiana. El modelo de aprendizaje automático de MALA se entrena para predecir la estructura electrónica basándose en esta vecindad atómica. Una ventaja significativa de MALA es la capacidad de su modelo de aprendizaje automático para ser independiente del tamaño del sistema, lo que permite entrenarlo con datos de sistemas pequeños y desplegarlo a cualquier escala.
En su publicación, el equipo de investigadores demostró la notable eficacia de esta estrategia. En comparación con los algoritmos convencionales, lograron una aceleración de más de 1.000 veces para sistemas de menor tamaño, de hasta unos pocos miles de átomos. Además, el equipo demostró la capacidad de MALA para realizar con precisión cálculos de estructura electrónica a gran escala, con más de 100.000 átomos. Cabe destacar que este logro se consiguió con un esfuerzo computacional modesto, lo que revela las limitaciones de los códigos DFT convencionales.
Attila Cangi, Jefe en funciones del Departamento de Materia en Condiciones Extremas del CASUS, explica: "A medida que aumenta el tamaño del sistema y hay más átomos implicados, los cálculos de DFT se vuelven poco prácticos, mientras que la ventaja de velocidad de MALA sigue creciendo. El principal avance de MALA reside en su capacidad para operar en entornos atómicos locales, lo que permite realizar predicciones numéricas precisas que se ven mínimamente afectadas por el tamaño del sistema. Este logro revolucionario abre posibilidades computacionales que antes se consideraban inalcanzables".
Se espera un impulso a la investigación aplicada
Cangi pretende ampliar los límites de los cálculos de estructuras electrónicas aprovechando el aprendizaje automático: "Prevemos que MALA desencadenará una transformación en los cálculos de estructuras electrónicas, ya que ahora disponemos de un método para simular sistemas significativamente mayores a una velocidad sin precedentes. En el futuro, los investigadores podrán abordar un amplio abanico de retos sociales partiendo de una base significativamente mejorada, como el desarrollo de nuevas vacunas y materiales novedosos para el almacenamiento de energía, la realización de simulaciones a gran escala de dispositivos semiconductores, el estudio de defectos materiales y la exploración de reacciones químicas para convertir el gas atmosférico de efecto invernadero dióxido de carbono en minerales inocuos para el clima."
Además, el enfoque de MALA es especialmente adecuado para la computación de alto rendimiento (HPC). A medida que crece el tamaño del sistema, MALA permite el procesamiento independiente en la malla computacional que utiliza, aprovechando eficazmente los recursos de HPC, en particular las unidades de procesamiento gráfico. Siva Rajamanickam, científico de plantilla y experto en computación paralela de los Laboratorios Nacionales Sandia, explica: "El algoritmo de MALA para el cálculo de estructuras electrónicas se adapta bien a los modernos sistemas HPC con aceleradores distribuidos. La capacidad de descomponer el trabajo y ejecutar en paralelo diferentes puntos de la malla en distintos aceleradores hace de MALA un complemento ideal para el aprendizaje automático escalable en recursos HPC, lo que conduce a una velocidad y eficiencia sin precedentes en los cálculos de estructuras electrónicas."
Además de los socios desarrolladores HZDR y Sandia National Laboratories, MALA ya es empleado por instituciones y empresas como el Georgia Institute of Technology, la North Carolina A&T State University, Sambanova Systems Inc. y Nvidia Corp.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original



Más noticias del departamento ciencias




































