La inteligencia artificial ayuda a explorar las fronteras de la química
El aprendizaje automático ayuda a simular la dinámica molecular reactiva para la investigación y el descubrimiento
La capacidad de simular el comportamiento de los sistemas a nivel atómico representa una poderosa herramienta para todo, desde el diseño de fármacos hasta el descubrimiento de materiales. Un equipo dirigido por investigadores del Laboratorio Nacional de Los Álamos ha desarrollado potenciales interatómicos de aprendizaje automático que predicen las energías moleculares y las fuerzas que actúan sobre los átomos, permitiendo simulaciones que ahorran tiempo y dinero en comparación con los métodos computacionales existentes.
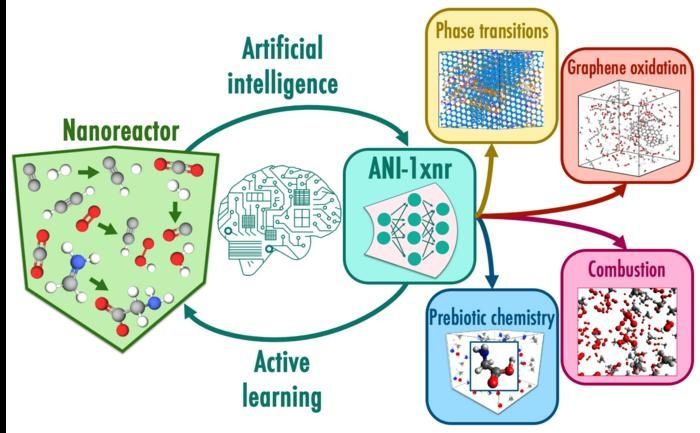
En este flujo de trabajo, las simulaciones de nanoreactores muestrean automáticamente el espacio químico reactivo sin depender de la intuición humana. El nanoreactor es una clase especial de simulaciones atomísticas en las que las reacciones químicas se inducen mediante la colisión de moléculas a altas velocidades. El aprendizaje activo utiliza el potencial de aprendizaje automático, ANI-1xnr, para dirigir la dinámica del nanoreactor y subseleccionar estructuras con grandes incertidumbres. Casos prácticos como las transiciones de fase de la combustión de carbono y metano ponen a prueba la generalidad del modelo resultante, ANI-1xnr.
Los Alamos National Laboratory
Según Benjamin Nebgen, físico químico de Los Álamos y coautor de un reciente artículo de Nature Chemistry en el que se describe el trabajo, "los potenciales de aprendizaje automático ofrecen cada vez más una alternativa eficaz a las simulaciones costosas desde el punto de vista computacional que intentan representar sistemas físicos complejos a escala atómica". "Un potencial interatómico de aprendizaje automático reactivo general, aplicable a una amplia gama de química reactiva sin necesidad de reajustes, beneficiará enormemente a la química y la ciencia de materiales".
Cerrar la brecha de las simulaciones eficaces
La construcción de simulaciones eficaces de dinámica molecular en química se realiza tradicionalmente con modelos computacionales basados en la física, incluidos los campos de fuerza clásicos o la mecánica cuántica. Aunque los modelos de mecánica cuántica son precisos y de aplicación general, resultan extremadamente caros desde el punto de vista computacional. Por el contrario, los campos de fuerza clásicos son eficientes desde el punto de vista computacional, pero su precisión es relativamente baja y sólo son aplicables a una gama limitada de sistemas. ANI-1xnr, el modelo de aprendizaje automático transformacional del equipo, salva la brecha de velocidad, precisión y generalidad que ha existido en química durante muchas décadas. (El aprendizaje automático es una aplicación de la inteligencia artificial en la que los programas informáticos "aprenden" mediante entrenamiento).
ANI-1xnr representa el primer potencial interatómico reactivo de aprendizaje automático lo suficientemente general -puede aplicarse a muchos sistemas químicos diferentes- como para competir con los modelos computacionales basados en la física a la hora de realizar simulaciones atomísticas reactivas a gran escala. ANI-1xnr se desarrolló utilizando un flujo de trabajo automatizado que realizaba simulaciones de dinámica molecular reactiva sobre una amplia gama de sistemas químicos que contenían elementos de carbono, hidrógeno, nitrógeno y oxígeno.
ANI-1xnr demostró ser capaz de estudiar una amplia gama de sistemas, desde las transiciones de fase del carbono hasta la combustión y la química prebiótica. El equipo validó las simulaciones comparándolas con resultados de experimentos y de técnicas computacionales convencionales.
Un potencial interatómico transformador
"ANI-1xnr no requiere conocimientos especializados ni reajustes para cada nuevo caso de uso, lo que permite a científicos de ámbitos muy diversos estudiar química desconocida", afirma Richard Messerly, científico computacional de Los Álamos y coautor del artículo. "La aplicabilidad general de ANI-1xnr es transformadora, ya que representa un paso significativo hacia la sustitución de las técnicas de modelado utilizadas desde hace tiempo para estudiar la química reactiva a escala".
El conjunto de datos utilizado por el equipo y el código ANI-1xnr se han puesto a disposición pública de la comunidad investigadora.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Shuhao Zhang, Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Olexandr Isayev, Nicholas Lubbers, Richard A. Messerly, Justin S. Smith; "Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential"; Nature Chemistry, 2024-3-7

Más noticias del departamento investigación y desarrollo





















































