Una brújula "molecular" muestra el camino para reducir los ensayos con animales
Científicos desarrollan una herramienta informática inteligente para evaluar los riesgos químicos
Anuncios



En los últimos años, los métodos de aprendizaje automático han cobrado cada vez más importancia para la evaluación del riesgo de los compuestos químicos. Sin embargo, también son una "caja negra" debido a la falta de trazabilidad y transparencia, lo que genera escepticismo entre los expertos y las autoridades reguladoras. Para aumentar la confianza en estos modelos, investigadores de la Universidad de Viena han identificado las áreas en las que presentan puntos débiles. Para ello han desarrollado una innovadora herramienta informática ("MolCompass").
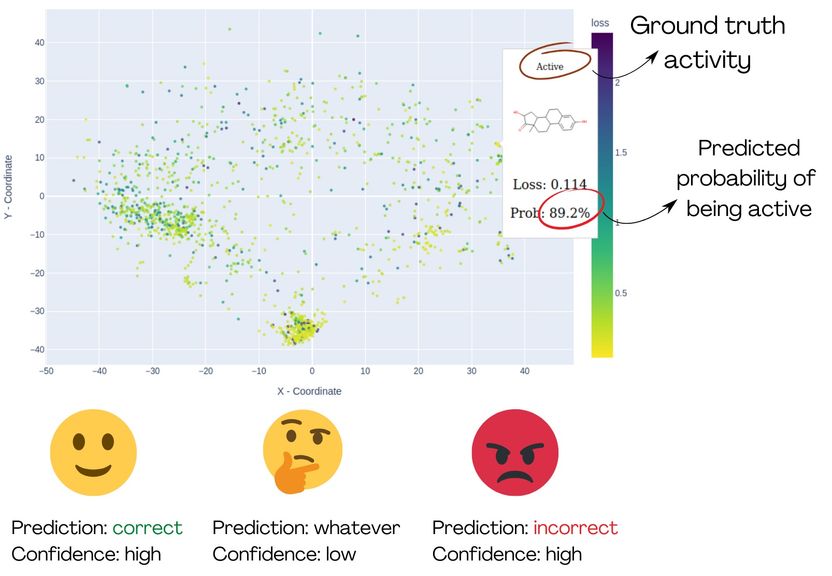
La demostración de MolCompass ilustra cómo los toxicólogos informáticos pueden identificar las zonas relevantes del espacio químico. Gracias a este programa, los toxicólogos pueden localizar regiones en las que el modelo analizado predice incorrectamente la actividad con un alto grado de certeza.
Sergey Sosnin
Durante muchas décadas, los nuevos fármacos y productos químicos agrícolas se probaban principalmente en animales. Estas pruebas son caras, plantean problemas éticos y a menudo no predicen con exactitud los efectos secundarios en humanos. En el marco del proyecto RISK-HUNT3R, financiado por la Unión Europea, científicos de la Universidad de Viena trabajan en el desarrollo de la próxima generación de métodos de evaluación de riesgos de nuevas sustancias sin animales. Los métodos asistidos por ordenador permiten ahora evaluar por ordenador los riesgos toxicológicos y ecológicos de nuevas sustancias químicas sin tener que sintetizar y probar los compuestos químicos. Pero queda una pregunta por responder: ¿Hasta qué punto son fiables estos modelos informáticos?
Predicciones fiables
Para investigar este problema con más detalle, Sergey Sosnin, científico principal del Grupo de Investigación Farmacoinformática de la Universidad de Viena, se centró en la clasificación binaria. En este caso, un modelo de aprendizaje automático proporciona una probabilidad del 0 % al 100 % que indica si un compuesto químico es activo o no (por ejemplo, tóxico o no tóxico, bioacumulativo o no bioacumulativo, aglutinante o no aglutinante de una proteína humana específica). Esta probabilidad refleja la confianza del modelo en su predicción. Idealmente, el modelo sólo debería dar valores cercanos al 0% (inactivo seguro) o al 100% (activo seguro) para predicciones correctas. Si el modelo es incierto y da una puntuación de confianza del 51%, por ejemplo, estas predicciones deben descartarse y utilizar métodos alternativos de evaluación de riesgos.
Sin embargo, el problema surge cuando el modelo da predicciones incorrectas con altas probabilidades: "Este es el verdadero escenario de pesadilla para los toxicólogos", dice Sergey Sosnin. "Si un modelo predice que un compuesto no es tóxico con un 99% de certeza, pero el compuesto es realmente tóxico, no hay forma de saber que algo ha ido mal". La única solución es identificar de antemano las zonas del espacio químico -es decir, las posibles clases de compuestos orgánicos- en las que el modelo tiene "puntos ciegos" y evitarlos. Esto exige que los investigadores que evalúan el modelo comprueben individualmente los resultados previstos para miles de compuestos químicos, una tarea tediosa y propensa a errores.
Superar este importante obstáculo
"Para ayudar a estos investigadores", prosigue Sosnin, "desarrollamos herramientas gráficas interactivas que proyectan los compuestos químicos en un plano 2D, similar a los mapas geográficos. Utilizando colores, resaltamos los compuestos que se predijeron incorrectamente con alta fiabilidad, permitiendo a los usuarios identificarlos como grupos de puntos rojos.
El mapa es interactivo y permite a los usuarios examinar el espacio químico y explorar las áreas de interés" La metodología se puso a prueba utilizando un modelo de unión al receptor de estrógenos.
Tras analizar visualmente el espacio químico, quedó claro que el modelo funciona bien, por ejemplo, para los esteroides y los bifenilos policlorados, pero falla por completo para los compuestos pequeños y no cíclicos, por lo que no debe utilizarse para ellos. El software desarrollado en este proyecto está a disposición gratuita de la comunidad científica en GitHub. Sergey Sosnin espera que MolCompass ayude a químicos y toxicólogos a comprender mejor las limitaciones de la modelización informática. Este estudio es un paso hacia un futuro en el que las pruebas con animales ya no serán necesarias y el único lugar de trabajo de los toxicólogos será un escritorio con un ordenador.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Alemán se puede encontrar aquí.
Publicación original



Más noticias del departamento ciencias




































